Dipolar relaxation¶
The system of interest here is an ensemble of identical spins characterized by a gyromagnetic ratio \(\gamma_I\) and spin quantum number \(I\). For \(^{1} \text{H}\), the most abundant isotope of hydrogen, \(I = 1/2\) and \(\gamma_I = 26.752\) rad/T/s. For \(^{13} \text{C}\), a natural and stable isotope of carbon, \(I = 1/2\) and \(\gamma_I = 6.728\) rad/T/s [25].
One assumption behind the theory presented here is that the cross-correlation terms can be neglected, see Ref. [4].
When the spin-lattice relaxation is dominated by fluctuations of the magnetic dipole-dipole interaction, as is the case for protons in molecular systems, the rates \(R_1 (\omega)\) and \(R_2 (\omega)\) are related to the spectral densities \(J^{(m)}(\omega)\) of these fluctuations via the Bloembergen-Purcell-Pound (BPP) equations [26]:
where
where \(\mu_0\) is the vacuum permeability, and \(\hbar\) the Planck constant (divided by \(2 \pi\)). The constant \(K\) has the units of \(\text{m}^6/\text{s}^2\). The spectral densities \(J^{(m)} (\omega)\) in Eq. (1) can be obtained as the Fourier transform of the autocorrelation functions \(G^{(m)}(\tau)\)
The spectral densities are a measure of the distribution of the fluctuations of \(G^{(m)}(\tau)\) among different frequencies, so they provide information on the distribution of the power available for causing spin transitions among different frequencies. The autocorrelation functions \(G^{(m)}(\tau)\) read
where \(F_2^{(m)}\) are some complex functions of the vector \(\textbf{r}_{ij}\) between the spin pairs, with norm \(r_{ij}\) and orientation \(\Omega_{ij}\) with respect to a reference applied magnetic field, assumed to be in the \(\textbf{e}_z\) direction. The functions \(F_2^{(m)}\) read
where \(Y_2^{(m)}\) are normalized spherical harmonics and \(\alpha_0^2 = 16 \pi /5\), \(\alpha_1^2 = 8 \pi /15\), and \(\alpha_2^2 = 32 \pi / 15\). Therefore, one can write for the correlation functions:
where \(N\) is the number of spins.
Intra/inter contributions¶
Intra-molecular and inter-molecular contributions to \(R_1\) and \(R_2\) can be extracted separately, by splitting the correlation functions as:
where \(j \in M_i\) and \(j \notin M_i\) refer to summation on spin from the same molecule as \(i\), and from different molecules as \(i\), respectively.
Intra-molecular relaxation is usually attributed to the rotational motion of the molecules, and inter-molecular relaxation to their translational motion. This is only an assumption that simplify the interpretation of results, and can lead to error [3].
Isotropic system¶
For isotropic systems, the correlation functions are proportional to each others, and only \(G^{(0)} (t)\) needs to be calculated.
In that case, \(G^{(0)} = 6 G^{(1)}\), and \(G^{(0)} = 6 / 4 G^{(2)}\) [19], and the spectrums can be calculated as:
which require less computational time and less memory to achieve, as only
needs to be evaluated. One can check the validity of the relation \(G^{(0)} = 6 G^{(1)} = 6 / 4 G^{(2)}\) on a simple bulk water system with 4000 molecules, similarly to what was done in Ref. [19] with glycerol.
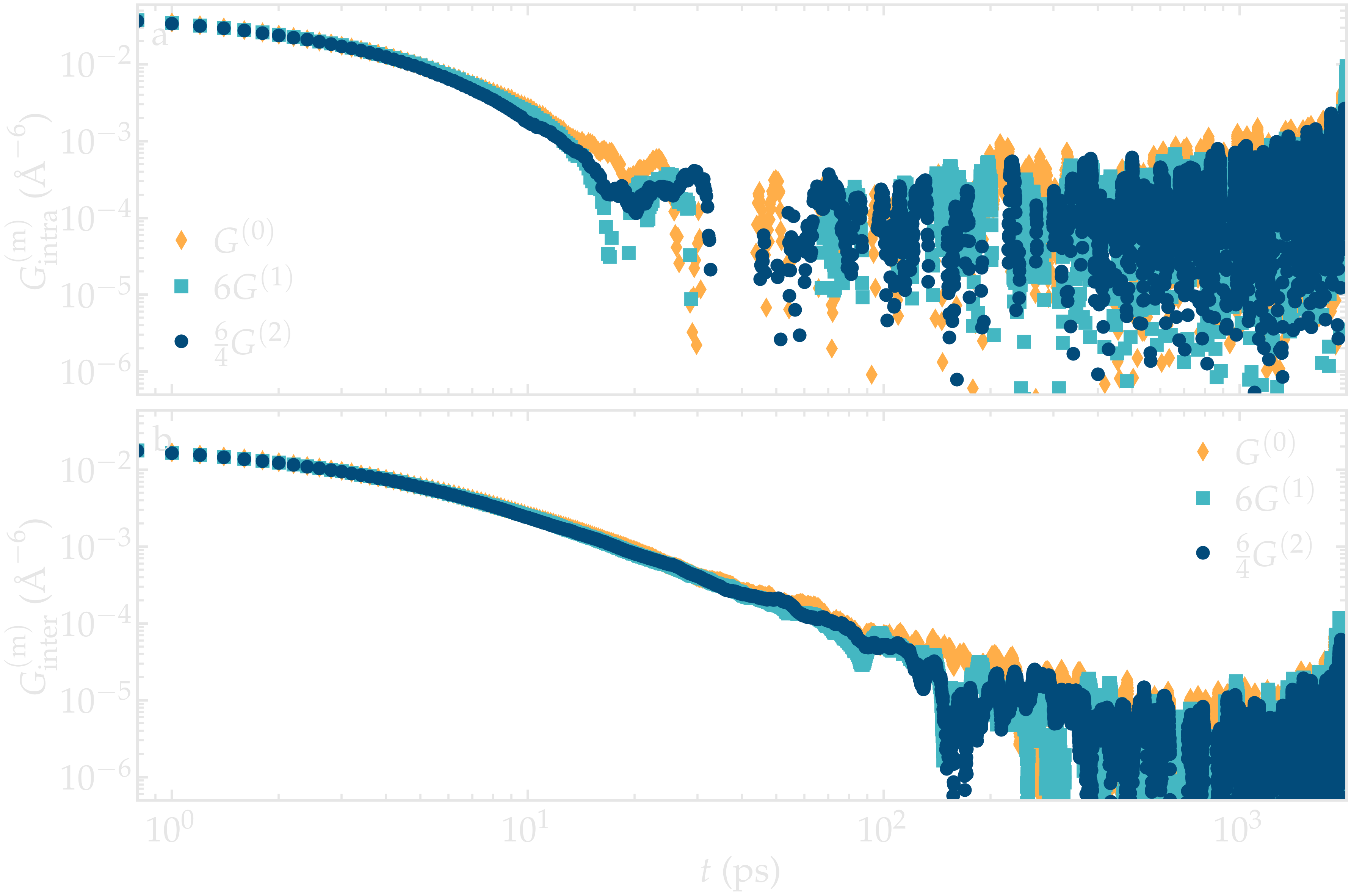
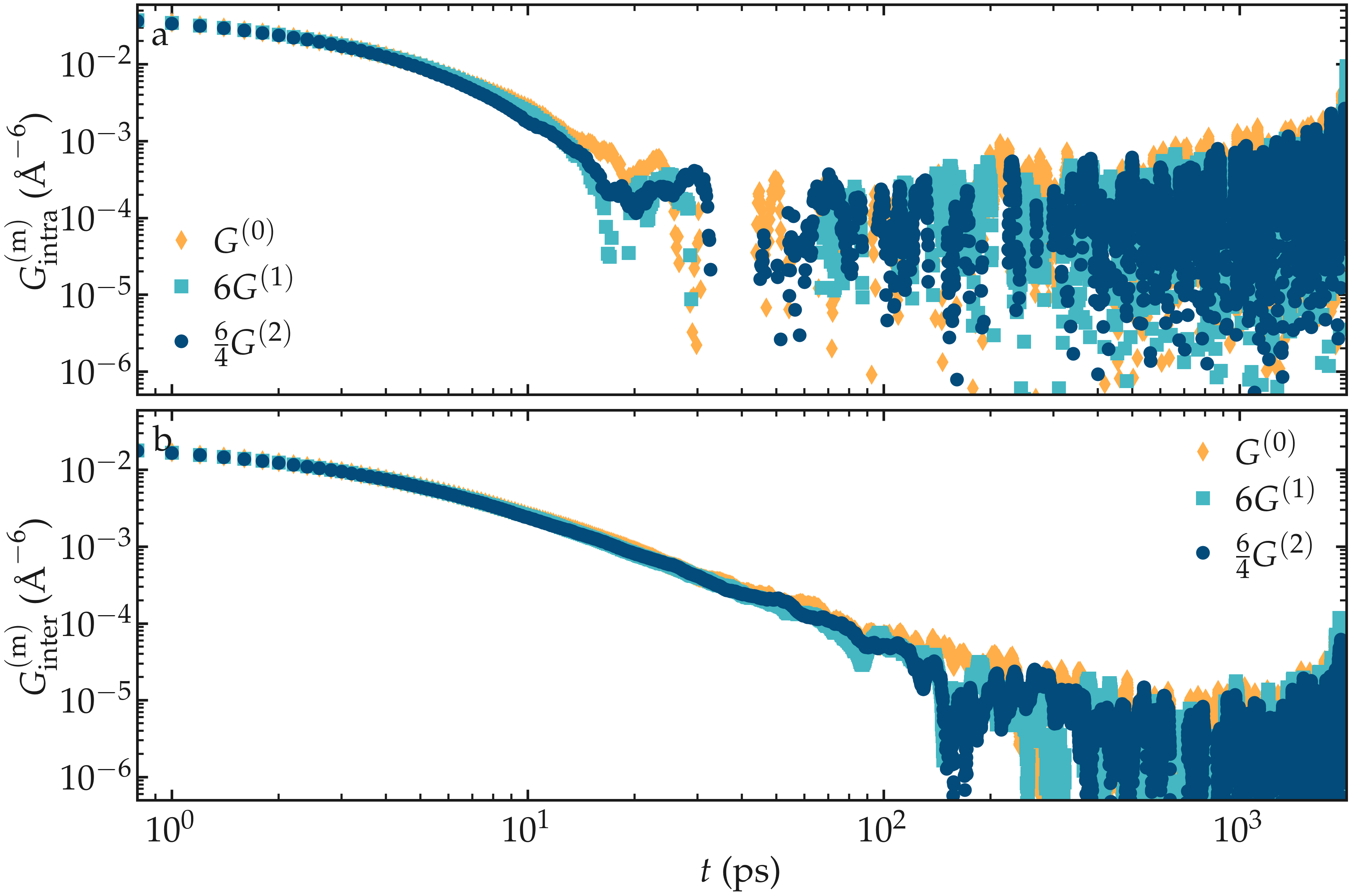
Figure: Validity of the relation \(G^{(0)} = 6 G^{(1)} = 6 / 4 G^{(2)}\) on a bulk water system.